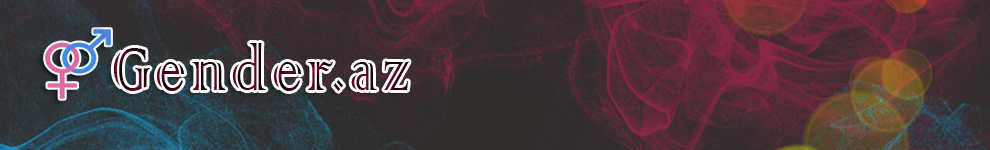
Bioloji Təbabət Klinikasının nəzdində olan Afgen Genetik Diaqnoz Mərkəzinin növbəti uğuru olan Pontoserebellar hipoplazi tip 6 xəstəliyi ilə əlaqəli daha əvvəl klinik ədəbiyata daxil olmamış yeni mutasiyadan söz açacağıq.
Pasiyentlərin həm genetik konsultasiyasını, həm dəbiyoinformatik data analizlərini həyata keçirdən İstanbul Universitetinin məzunu, genetikmütəxəssis İlahə Musayevanın təqdimatında həmin ailə hekayəsi ilə tanış olun.
Pontoserebellar hipoplazi, beynin inkişafına təsir edən xəstəlik qrupudur. Pontoserebellar hipoplazi beynin digər hissələrinin böyüməsinin pozulmasına səbəb olur və bu da mikrosefaliyaya səbəb olur.
Mikrosefaliya vəziyyəti adətən doğuşda görünmür, ancaq beynin böyüməsi körpəlikdə və erkən uşaqlıqda yavaş olmağa davam etdiyi üçün nəzərə çarpır. Araşdırmacılar pontoserebellar hipoplaziyanın ən az 10 növü olduğunu qeyd edirlər. Bütün növlər beyin inkişafının pozulması, ümumi inkişafın ləngiməsi, hərəkət problemləri və əqli gerilik ilə xarakterizə olunur.
Beyin anomaliyaları ümumiyyətlə doğuş zamanı olur və bəzi hallarda doğumdan əvvəl aşkar edilə bilər.
Pontoserebellar hipoplaziyası olan bir çox uşaq yalnız körpəlik dövrünə qədər yaşaya bilirlər.
Xəstəliyin irsiyyət tipi autozomal resessivdir.
1-ci dərəcədən qohum evliliyi etmiş ailə (bibi qızı-dayı oğlu) Afgen Genetik Diaqnoz Mərkəzinə genetik konsultasiya üçün yaxınlaşdığında çoxsaylı inkişaf qüsuru ilə artıq iki körpəsini (Hamiləliyin 19.cu həftəsində beyincik inkişafının durması) itirmişdi və bir sonraki hamiləliyin artıq planlı bir şəkildə prenatal diaqnostika ilə həyata keçirdilməsini istəyirdi.
3-cü hamiləlik süni doğuşla sonlandırıldıqdan sonra körpədən də nümunə götürüldü.
Differensial diaqnostika və dəqiq diaqnoz məqsədilə ata və ananın hər ikisinə WES (bütün ekzom sekanslama) analizinin edilməsi tövsiyə edildi. Pasiyentlərin WES datası dəqiq biyoinformatik analiz programları vasitəsi ilə genetik mütəxəssisimiz Ilahə Musayeva tərəfindən incələndikdən
sonra ata və ananın hər ikisində Pontoserebellar hipoplaziya tip 6 xəstəliyi ilə əlaqəli RARS2 genində mutasiya aşkarlandı. Körpədən əldə edilmiş DNT nümunəsindən mikroarray (aCGH) analizi edildi və analizin nəticəsində kliniki vəziyyətlə əlaqəli xromosomal kopya sayı dəyişikliyi aşkarlanmadı.
Anada RARS2 genində klinik olaraq tanımlı olmayan patojenik c. C733T (p. R245W) (DM)variantı heterozigot vəziyyətdə aşkarlandı. Bəhsi keçən varyant yanlış anlamlı varyantların patojeniteleri haqqında təxmin dəyəri verən REVEL və Mutation Taster programlarından keçirdilmişdir.
REVEL in silico təxmin programı bəhsi keçən varyant üçün verdiyi dəyər
sayəsində (REVEL: 6,88240540,87530822,G,A,R,W,0.575) varyant haqqında “zərərli, xəstəlik səbəbi olabilər” ehtimalı bildirildi. Eyni zamanda Mutation taster veritabanında da bu varyant haqqında DM (disease-causing) olduğu belirtildi. Sanger konfirmasyon analizi edildi.
Atada RARS2 genində klinik olaraq tanımlı patojenik c.633_636del:p.E212Qfs*7 (DM) variantı heterozigot vəziyyətdə aşkarlandı. Bəhsi keçən varyant haqqında Mutation taster və ClinVar
veritabanlarında “pathogenic” olduğu belirtildi. Sanger konfirmasyon analizi edildi.
Ana və atanın bəhsi keçən xəstəlik üçün daşıyıcı olduğu müəyyənləşdikdən sonra süni doğuş edilmiş körpədən əldə edilən DNT’dən bəhsi keçən mutasiyalar üçün segregasiya analizi edildi və körpədə hər iki mutasiya baxımından kompound-heterozigot vəziyyətinin olduğu aşkarlandı.
Ailəyə düzgün genetik danışma və prenatal diaqnostika haqqında məlumat verildi. Bu çalışmanıntibb aləmi üçün dəyərli olmasının səbəbi həm xəstəliyin bu tipinin olduca nadir olması, həm də aşkarlanan mutasiyalardan birinin daha öncə klinik ədəbiyyatda yer almamış olmasıdır.\\Medicina










 Ölkəmizdə psixi xəstələrin sayı sürətlə artır - Şok
Ölkəmizdə psixi xəstələrin sayı sürətlə artır - Şok “Məni vurdu, bildirdi ki, evdə desən, qiymətini kəsəcəyəm” - Video
“Məni vurdu, bildirdi ki, evdə desən, qiymətini kəsəcəyəm” - Video 40 yaşdan sonra əksər insanların damarı tutulub- Diqqət
40 yaşdan sonra əksər insanların damarı tutulub- Diqqət Mübahisəli ağacla bağlı qəribə qərar verildi
Mübahisəli ağacla bağlı qəribə qərar verildi Avtosığorta qaydalarında YENİLİK
Avtosığorta qaydalarında YENİLİK Abel Məhərrəmova vəzifə VERİLDİ
Abel Məhərrəmova vəzifə VERİLDİ "BakıKart"ın qiyməti bahalaşdı
"BakıKart"ın qiyməti bahalaşdı Qaz kəmərində PARTLAYIŞ - bu ölkədə
Qaz kəmərində PARTLAYIŞ - bu ölkədə Milli Məclisin növbədənkənar sessiyası - Deputatlar bunun üçün toplaşır
Milli Məclisin növbədənkənar sessiyası - Deputatlar bunun üçün toplaşır Süni intellekt peyvənd yaratdı, insanlar üzərində sınaqdan keçirdi: Nə baş verdi?
Süni intellekt peyvənd yaratdı, insanlar üzərində sınaqdan keçirdi: Nə baş verdi?



